 论文排版软件
论文排版软件大家好,今天为大家介绍一篇发表在JACS上的文章,“Self-Improving Photosensitizer Discovery System via Bayesian Search with First-Principle Simulations”。在这篇文章中,作者首次演示如何通过量子力学和贝叶斯优化的人工智能(AI)学习方法,应用于高性能光敏剂(PSs)的自主更新探索,此系统可以准确预测材料的性能,使下一代光敏剂材料的发现更为容易。这样的系统在单线态-三重态分裂的平均绝对误差为0.090 eV,此极小的误差值使其具有对高性能PSs的搜索能力。作者从超过700万个分子中归纳出5357个潜在的高性能PSs,且其中四个PSs被进一步合成并证实其性能优于现有的商业光敏剂。

背景介绍
人工智能(AI)算法和硬件的快速发展,逐渐改变了人们对材料搜索的思维方式,通过引进AI进行高通量筛选,辅以量子计算(如第一性原理计算)评估一种材料是否能达到所期望的性能,可以加快新材料的发现与设计。光敏剂(PSs)是一种可以吸收光并产生单线态氧、活性氮和自由基的分子。通过泛函理论(TD-DFT)方法对ΔEST计算,可以预筛选出有潜力的PS。然而,到目前为止, PS的发现都局限于”耗时”的 DFT和 TD-DFT计算且分子的结构建立都依赖于经验。在本文中,作者首先从费米黄金法则推导方程式,确定了高效率PS设计的原则。接下来,通过AI分子生成算法,构建了12015个随机选择的分子模型,这些结构形成了初始数据集,并用于模型训练并生成预测模型。
设计思路
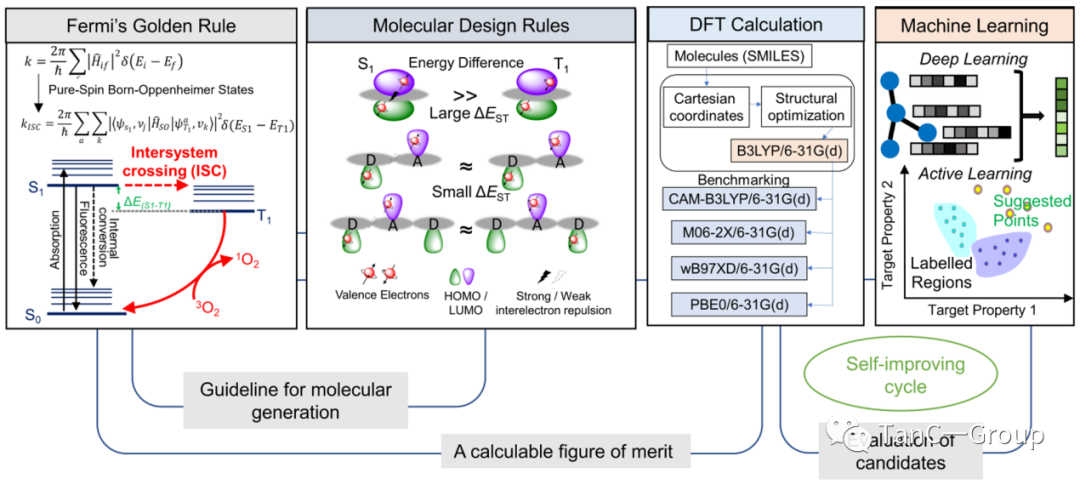
图1.基于量子机制的PS发现系统
首先,将雅布隆斯基图(Jablonski Diagram)连接到机器学习是关键步骤。Jablonski图中单线态氧产生的关键是从单线态到三重态的交叉能差(ISC),对高性能PSs的要求是高ISC率(kisc),而根据费米黄金定律可知kisc与S1−T1间隙成反比。因此,降低ΔEST(低于0.3 ~ 0.5 eV)是产生高效PSs的关键要求之一。根据以上设计原理,作者开发了供体-受体(DA)和供体-受体-供体(DAD)的分子设计方案,这是因为S1和T1之间的能量差来源于具有相反自旋的S1的价电子,从而导致电子排斥增加了分子的总能量。因此,降低ΔEST是为了减轻S1态的电子斥力。由于电子斥力和价电子间的距离成反比,分离最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)的分布是有效降低ΔEST的关键。
接着,作者进行分子空间的生成和初步预测。如图2a所示,作者先以一个抽象的分子图作为神经网络的输入信息,再将有用的信息,如原子数量,编码到这个输入表示中,接着生成一个长条图作为分子指纹。如图2b,c所示,对两个初始DA和DAD进行ΔEST预测,从统计指标的平均绝对误差(MAE)来看,机器学习预测的ΔEST值与通过量子化学计算的ΔEST值的结果非常接近。
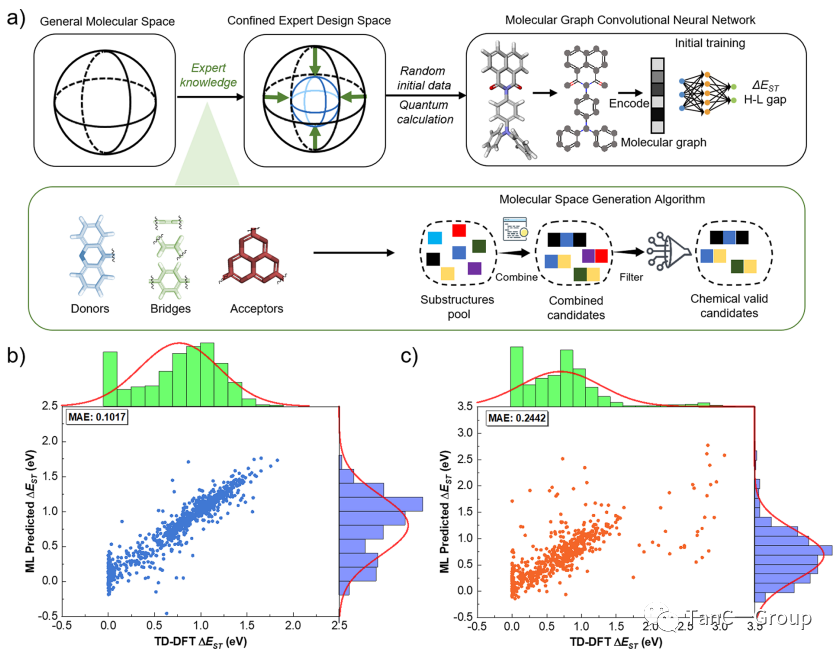
图2. DA 和 DAD PSs 的分子空间生成算法示意图,平均绝对误差 (MAE) 和 ΔEST 预测分布
实验结果与讨论
下一步,作者通过贝叶斯优化来建立完善的AI循环。如图3a,b所示,先采用此分子预测模型计算出贝叶斯优化(BO)评分和预期改善评分 (EI),接着根据此结果使机器在每个周期进行主动学习。图3c,d显示如何从MAE的变化模型中找到ΔEST< 0.2 eV 的潜在的PSs,每一回机器学习的结果会得到ΔEST < 0.2 eV的PSs占比,随着主动学习周期的增加,可以看出DA(图3c)和DAD(图3d)的发现比例有上升。
如图3e所示,初始随机训练的数据均匀分布在整个设计空间,保证了模型对整个设计的通用性。循环1−8(图3e中的DA模型)更为明显,大多数建议的分子形成了几个显着的集群,只有零星几点是分离的,可以将其忽略。
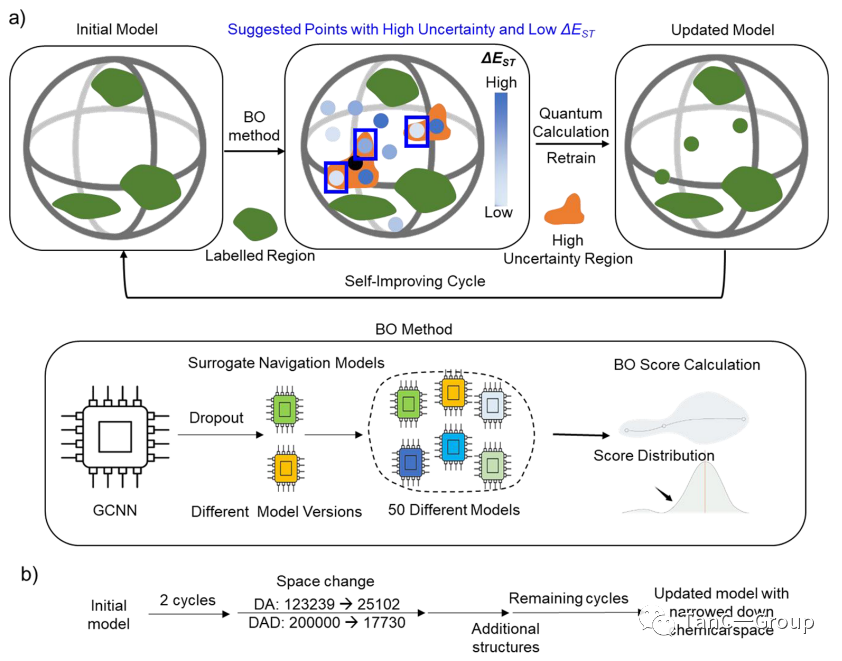
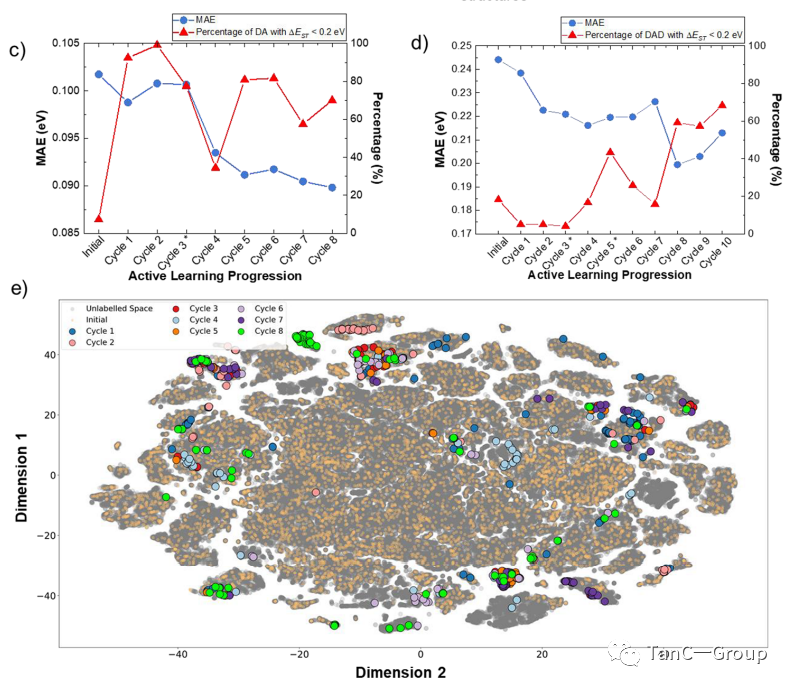
图3.DA 和 DAD PS 的贝叶斯优化主动学习示意图
通过以上计算后,有潜力的PS被缩减到人类可处理的决策数量,作者对可能的候选PS进一步评估其性质,如HOMO – LUMO差距,结构新颖性,分子量和合成可行性(图4a,b)。从预测结果来看,四个候选PS的HOMO – LUMO差距范围均在从1.33到2.51 eV(图中红色到蓝色),作者将其合成出并测试光谱来验证此系统的推断结果(图4c,d)。
被选择的分子如图4d所示,分别为含有吩噻嗪,吖啶酮,苯恶嗪,三苯胺的给体和含有2,2′-(蒽-9,10-二亚基)二氨基腈、萘酚[2,3-c][1,2,5]噻二唑,蒽-9,10-二酮和二苯[f,h]喹恶啉-2,3-二羰基腈的受体。作者首先对这四种材料的光学性质进行了表征,通过测量化合物的吸收和发射光谱(图4e,f),发现系统的预测与实验结果一致,接下来作者使用ABDA [9,10-蒽二酰-双(亚甲基)二丙二酸]来测试四个分子的PS单线态氧的生成效率(图4g,h),并和三种常用的商用PSs,包含chlorin e6 (Ce6)、亚甲基蓝(MB)和玫瑰红(RB)在相同条件下进行比较。令人印象深刻的是,4种化合物表现出的ABDA降解率皆可与三个商业PS相媲美。
为了排除分子对光吸收差异的影响,作者将降解率除以不同化合物在可见范围内的吸收值(图4 h),结果同样显示所有化合物的单线态氧产生效率都相当于或高于这三个商业PS,更值得注意的是化合物3的降解率最高,分别是Ce6、MB和RB的5倍、3倍和2.5倍。这些结果不仅验证了此搜索系统在新分子的光学预测能力(图4i),也证明了其对下一代PS的搜索能力。
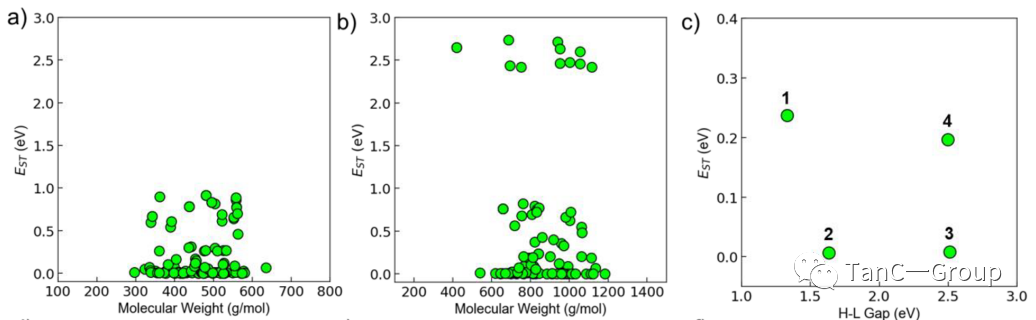
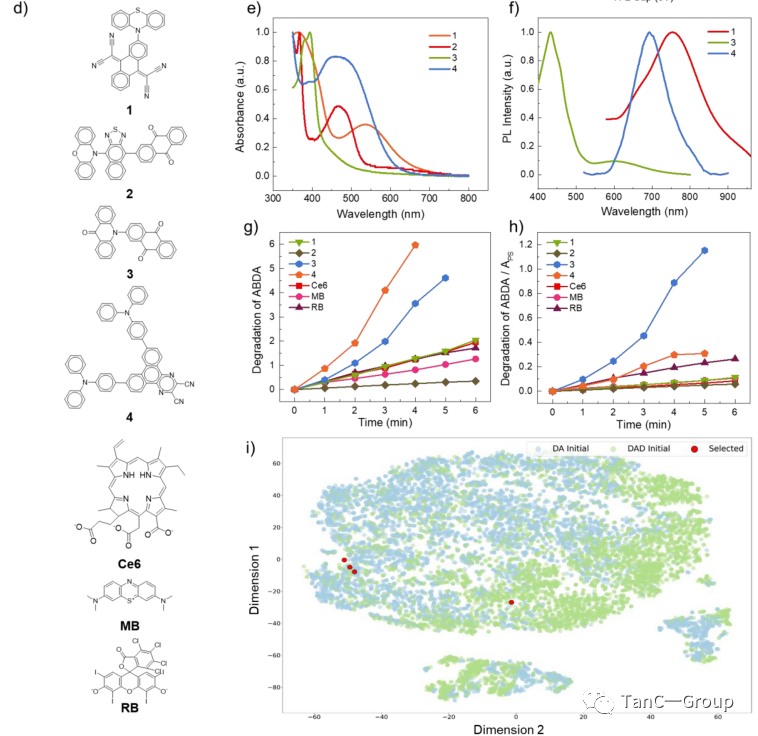
图4. 1-4分子的实验数据
结语
综上所述,该课题组开发出一种PS发现系统,具有很强大的分子搜索能力和高精度的分子性质预测能力。ΔEST的预测和主动学习的结合,可以形成一个AI的学习循环,推进未来在材料领域的新发现。此PS搜索系统成功的关键在于对PS性质的基本理解足够深刻,是第一个基于原理设计的PS发现系统。本文不仅有评估PS效率的模拟结果,也构建一个非常有前景的PS分子文库。总计14164个PSs数据在整个过程中被标记,且经AI自行改进,ΔEST误差仅为0.090eV,筛选的结果共有2478个PSs的ΔEST< 0.2eV,2879个PSs光谱覆盖整个可见范围。综合上述,此系统对未来利用AI进行材料探勘做出了完美的演示,并成功筛选出四个有前途的PSs结构,指明了未来光动力治疗的发展方向。

本文作者|JW
审稿|Tulip,鲁班一号
责任编辑|鲁班一号,海猫
原文引用|10.1021/jacs.1c08211
